Graham Muir1,2, Christian Schlötterer2*
1 Department of Applied Plant Science, The Queen's University
of Belfast,
2 Institut für Tierzucht und Genetik,
Veterinärmedizinsche Universität Wien,
*Corresponding author: Email: Christian.Schloetterer@vu-wien.ac.at
Introduction
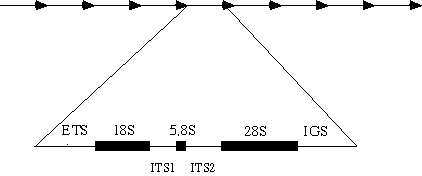
Ribosomal DNA (rDNA) codes for the RNA component of the ribosome. The rDNA is a multigene family with nuclear copies in eukaryotes arranged in tandem arrays (Figure 1). They are organised in nucleolus organiser regions (NORs), potentially at more than one chromosomal location. Each unit within a single array consists of the genes coding for the small and large rRNA subunits (18S and 28S). The 5.8S nuclear rDNA gene lies embedded between these genes but separated by two internal transcribed spacers: ITS1 and ITS2. The external transcribed spacer (ETS) and the intergenic spacer (IGS) separate the large and small subunit rDNAs. The copy numbers of 18S-5.8S-28S rRNA genes in diploid genomes of Quercus cerris, Q. ilex, Q. petraea, Q. pubescens and Q. robur have been estimated to be in the range of 1300-4000 (Zoldos et al. 1999).
Figure 1: Organisation of one rDNA array. Single repeat units (arrows) are tandemly organised. Each of them consists of the rRNA genes: 18S, 5.8S and 28S. Spacers separate these genes, namely the external transcribed spacer (ETS), the internal transcribed spacers (ITS 1 and ITS 2) and the intergenic spacer (IGS).
Different selective forces are acting on the rDNA region with the consequence of varying degrees of sequence conservation across single repeat units. Therefore, each part can be employed for specific phylogenetic questions across a broad taxonomic spectrum (Hills and Dixon 1991). The small subunit is highly conserved and has been used to shed light on deep evolutionary branches, e.g. for relationships between Archaebacteria and Eubacteria, while the more conserved domains within the 28S region have been used to cover evolutionary time through the Paleozoic and Mesozoic eras. The faster evolving ITS regions, however, have been employed for populations and congeneric phylogenies (e.g. Bayer et al. 1996). The smallest rDNA gene of the cluster, the 5.8S, is too short to provide a robust phylogenetic signal.
Why should we use the internal transcribed spacer (ITS) as a marker ?
PCR amplification of the ITS region has become a popular choice for phylogenetic analysis of closely related species and populations. This popularity stems from the derivation of universal primers located in the coding regions flanking the ITS. Both direct sequencing and cloning of PCR products can be used for ITS analysis. Their deployment depends on the hierarchical scale of the question. Direct sequencing generates a consensus sequence for phylogenetic analysis. For population questions, additional profit is gained from information among single repeat units. Hence, PCR products need to be cloned and sequenced.
Ribosomal DNA genes evolve cohesively within a single species and exhibit only limited sequence divergence between rDNA copies within single individuals (Arnheim et al. 1980). In contrast, comparisons between species show normal levels of sequence divergence. The combination of these two observations is referred to as concerted evolution (Dover 1982). The mechanisms driving concerted evolution are unequal crossing over and gene conversion. Irrespective of the exact mechanism, the degree of homogenisation is a result of the interplay between homogenisation mechanisms and mutation processes (Schlötterer and Tautz 1994). The rDNA clusters are frequently distributed on several chromosomes. A potential problem is understanding to what extent concerted evolution is obstructed by the different chromosomal locations of the arrays. The purpose of this study was to ask whether the ITS could be employed as a universal marker, using oaks as a model. Can the rDNA be treated as a single gene in Quercus petraea ?
Materials and Methods
DNA extraction
Total genomic DNA was extracted from leaves using Nucleon Phytopure (Amersham) according to manufacturer's instructions. The internal transcribed spacers (ITS1 and ITS2) and 5.8S of the rDNA repeat array were sequenced from a single Quercus petraea individual. The species was identified using morphology (Clapham et al. 1981) and microsatellite analysis (Muir et al., unpublished data).
PCR Amplification
Initially, universal primers 18S, TCG TAA CAA GGT TTC CG and 28S, GTT RGT TTC TTT TCC TC (Schlötterer et al. 1994) were used, but they amplified non-plant ITS sequences. Therefore, plant-specific primers were designed with the following sequences: 18S, CCT TMT CAT YTA GAG GAA GGA G and 28S, CCG CTT ATT KAT ATG CTT AAA. A 40 µl reaction was prepared with 100 ng of genomic DNA, 1.5 mM MgCl2, 2 mM dNTPs, 20 µM of each primer and 0.5 U Taq polymerase. The cycling profile consisted of an initial denaturation step of 3 min followed by 40 cycles of 60 seconds at 94°C, 60 seconds at 56°C and 90 seconds at 72°C, and no final extension. These PCR products were blunt-ended using Klenow DNA Polymerase I (GibcoBRL) and subsequently cloned.
Cloning and Sequencing
The standard 20 µl ligation mix contained 20-100 ng of phosphorylated PCR product, 100 ng M13 mp19 (Messing et al. 1977), 1 U T4 ligase (Promega) and 1x T4 ligation buffer (Promega). Ligation was carried out at 18°C overnight. Clones carrying inserts were identified with blue/white selection. Sequencing templates were prepared from overnight cultures of positive clones using standard protocols. Clones were sequenced using ABI Dye Terminator chemistry (Perkin Elmer) according to manufacturer's instructions and run on a Perkin Elmer ABI 377 automated sequencer.
Data Analyses
Sequences were edited in Sequence Navigator (Perkin Elmer) and aligned using CLUSTAL W (Thompson et al. 1994). The alignment was adjusted manually in SEQPUP and the phylogeny was computed using a Maximum Likelihood approach as implemented in PUZZLE 4.0 (Strimmer and von Haeseler 1996). The HKY (Hasegawa et al. 1985) model of base substitution with rate heterogeneity was used. The phylogenetic tree was displayed using TREEVIEW (Page 1996). One clone from Quercus cerris, a close relative of Q. petraea, was used as an outgroup. Tables of polymorphic sites were produced using SITES (Hey and Wakeley 1997).
Results
The use of universal primers for amplification from oak leaf material resulted in the preferential amplification of endosymbiont and endoparasitic ITS sequences. DNA preparations from natural populations are often contaminated with primitive eukaryotic DNA containing shorter ITS regions which may amplify more efficiently. Consequently, without prior sequence information, it is easy to misclassify the amplified ITS sequences.
Plant specific primers were designed by hand from a published alignment of 18S and 28S sequences (Schlötterer 1998). In response to polymorphisms in the alignment, wobbles were incorporated to recognise all plant ribosomal targets in principle. The primers amplified successfully and showed only one defined band on an agarose gel. To verify the origins of these PCR products, we conducted a GenBank BLAST search (Altschul et al. 1990) which showed significant sequence homology with other Quercus rDNA sequences.
Table 1 gives the polymorphic sites in the 5.8S (161 bp) and the ITS2 (228bp) for 13 Q. petraea clones and the single Q. cerris clone used as an outgroup. The surveyed clones showed an average pairwise distance of 0.0405 in the 5.8S coding region. No insertions or deletions of nucleotides (indels) were observed in this region (Table 1a). In contrast to those findings, in the ITS2, which is supposed to be evolving at a faster rate, the average pairwise distance among clones is 0.0707. The indels range from one to four base pairs (Table 1b). The nucleotide distribution is balanced in the 5.8S, while in the ITS2 the GC content is higher.
(a) position
11111 A = 25.0%
position 135567899 02346 C = 24.3%
position 4132407667 81361 G = 27.9%
190/3.2 GGGCAGGCCG CCGCG T = 22.8%
190/3.5 ---------- -----
190/1.9 ---------- -----
190/1.8 ---------- -----
190/2.7 ---------- -----
190/2.0 ---------- -----
190/1.6 ---------- -----
190/2.5 ---------- -----
190/3.0 AA-TG-TTT- ATA-A
190/1.3 AA-TG-TTT- TTA-A
190/1.7 --AT-A---A ---T-
190/3.6 --AT-A---A ---T-
190/3.1 --AT-A---A ---T-
Cerris7.6 --A--A--A- TT-T-
(b) position
11111111 1111111111 222222 A = 14.5%
position 111123344 6667777778 8900112345 5777778888 001112
C = 37.9%
position 5567897824 6890134590 5548374726 8167894579 010120
G = 27.6%
190/3.5 GC***GCGCG GTCCCCACCG GGGCGCCGCT CGAACGCCAA GTGGCA
T = 20.1%
190/2.7 ---------- ---------- ---------- ---------- ------
190/3.2 ---------- ---------- ---------- ---------- ------
190/2.0 ---------- ---------- ---------- ---------- ------
190/1.9 ---------- ---------- ---------- ---------- ------
190/1.6 ---------- ---------- ---------- ---------- ------
190/1.8 ---------- ---------- ---------- ---------- ------
190/2.5 ---------- ---------- ---------- ---------- ------
190/3.0 -*----TATA C****-G-T- AAATAT-T-C T-****TTGG **ATTC
190/1.3 -*---ATATA C****-G-T- AAATAT-T-C T-****TTGG **ATTC
190/1.7 A-CCAAT-TA C****T-T-T -AA--TTA-C TT****-TGG **-ATC
190/3.6 A-CCAAT-TA C****T-T-T -AA--TTA-C TT****-TGG **-ATC
190/3.1 A-CCAAT-TA C****T-T-T -AA--TTATC TT****-TGG **-ATC
Cerris7.6 --CCCA-A-- C****-GT-- -A--T--ATC T-****-T-G **---C
Table 1: Polymorphic sites between the surveyed
Quercus petraea clones (190): (a) 5.8S (161 bp) and (b) ITS2 (228
bp). The three divergent clades in Figure 1 are readily apparent in the
table. Nucleotide frequencies are also given. Invariant sites are marked
with a dash, asterisks refer to indels.
In light of the previous discussion on concerted evolution, it is worth restating what we expect to see for a rDNA phylogeny based on sequenced PCR clones. As a result of homogenisation by gene conversion and/or recombination, little if any nucleotide divergence is expected among single array units, regardless of whether they belong to one or several arrays, within a single Quercus petraea individual. In contrast to expectation, substantial sequence divergence of 4% and 7% in the 5.8S and ITS2, respectively, is apparent (Table 1). This suggests the presence of more than one rDNA array in single individuals. The phylogenetic tree based on the 5.8S and ITS2 further confirms this. Rather than displaying only one clade, derived from the homogeneous units of the array(s), the tree shows three clades, highly divergent and all co-occurring within one individual (Figure 2).
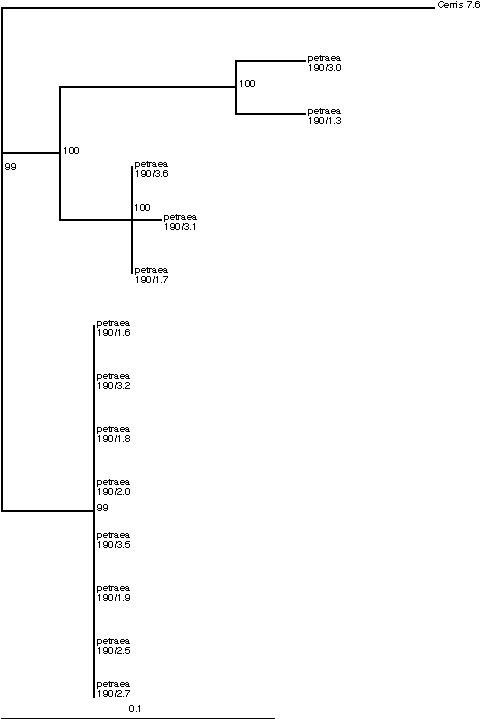
Figure 2: Maximum Likelihood tree of 5.8S (161 bp) and ITS2 (228 bp) PCR clones from a single Quercus petraea individual. 13 clones were sequenced from one PCR amplification. Supports are quartet puzzling values (Strimmer and von Haeseler 1996).
Discussion
The sequenced clones from a single Q. petraea individual reveal that there are at least three haplotypes within the genome. This indicates that there may be three independent loci (nucleolus organiser regions NORs). In Quercus petraea, the presence of NORs on independent chromosomes has also been documented using fluorescence in situ hybridisation (Zoldos et al. 1999). Consistent with these results are observations in other species, e.g. arbuscular mycorrhizal fungi (Hosny et al. (1999), platyhelminths (Carranza et al. 1996) and aphids (Fenton et al. 1998). The three clades are highly divergent and probably can be viewed as three independent phylogenetic entities rather than resulting from a low rate of concerted evolution. If incomplete concerted evolution were responsible for the observed differences between rDNA clades, then the same nucleotide distance between and within clades would be expected.
The presence of divergent clades will result in an ambiguous consensus sequence when sequencing PCR products directly for phylogenetic analysis. Similarly, for population studies, if members of any array are absent in the cloning, the phylogenetic analysis will be suspect. These results underline that the use of the ITS as a universal marker should be evaluated on a case by case basis. The use of ITS sequences as molecular marker relies on the mechanisms of concerted evolution to ensure the rDNA array is evolving as a single molecule. Because the three divergent clades found in this study most likely predate events at the population level, they cannot be used reliably to answer questions at this hierarchical level.
Conclusions
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. Journal of Molecular Biology 215(3): 403-410.
Arnheim N, Krystal M, Schmickel R, Wilson G, Ryder O, Zimmer E (1980) Molecular evidence for genetic exchanges among ribosomal genes on non-homologous chromosomes in man and apes. Proceedings of the National Academy of Sciences of the USA 77(12): 7323-7327.
Bayer RJ, Soltis DE, Soltis PS (1996) Phylogenetic inferences in Antennaria (Asteraceae: Gnaphalieae: Cassiniinae) based on sequences from nuclear ribosomal DNA internal transcribed spacers (ITS). American Journal of Botany 83(4): 516-527.
Carranza S, Giribet G, Ribera C, Baguñà J, Riutort M (1996) Evidence that two types of 18S rDNA coexist in the genome of Dugesia (Schmidtea) mediterranea (Platyhelminthes, Turbellaria, Tricladida). Molecular Biology and Evolution 13(6): 824-832.
Clapham AR, Tutin TG, Warburg EF (1981) Excursion Flora of the British Isles. Cambridge University Press.
Dover G (1982) Molecular drive: a cohesive mode of species evolution. Nature 299: 111-117.
Fenton B, Malloch G, Germa F (1998) A study of variation in rDNA ITS regions show that two haplotypes coexist within a single aphid genome. Genome 41: 337-345.
Hasegawa M, Kishino H, Yano K (1985) Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22: 160-174.
Hey J, Wakeley J (1997) A coalescent estimator of the population recombination rate. Genetics 145: 833-846.
Hillis DM, Dixon MT (1991) Ribosomal DNA: molecular evolution and phylogenetic inference. The Quarterly Review of Biology 66(4): 411-446.
Hosny M, Hijri M, Passerieux E, Dulieu H (1999) rDNA units are highly polymorphic in Scutellospora castanea (Glomales, Zygomycetes). Gene 226: 61-71.
Messing J, Gronenborn B, Muller-Hill B, Hofschneider PH (1977) Single strand filamentous DNA phage as a carrier for in vitro recombined DNA. Proceedings of the National Academy of Sciences of the USA 74: 3642-3646.
Page RDM (1996) TREEVIEW: An application to display phylogenetic trees on personal computers. Computer Applications in the Biosciences 12: 357-358.
Schlötterer C (1998) Ribosomal DNA Probes and Primers. In: Karp A, Isaac PG, Ingram DS (eds.). Molecular Tools for Screening Biodiversity. Chapman & Hall, London.
Schlötterer C, Hauser MT, von Haeseler A, Tautz D (1994) Comparative evolutionary analysis of rDNA ITS regions in Drosophila. Molecular Biology and Evolution 11(3): 513-522.
Schlötterer C, Tautz D (1994) Chromosomal homogeneity of Drosophila ribosomal DNA arrays suggests intrachromosomal exchanges drive concerted evolution. Current Biology 4: 777-783.
Strimmer K, von Haeseler A (1996) Quartet puzzling: a quartet maximum likelihood method for reconstructing tree topologies. Molecular Biology and Evolution 13: 964-969.
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Research 22: 4673-4680.
Zoldos V, Papes D, Cerbah M, Panaud O, Besendorfer V, Siljak-Yakovlev S (1999) Molecular-cytogenetic studies of ribosomal genes and heterochromatin reveal conserved genome organisation among 11 Quercus species. Theoretical and Applied Genetics 99: 969-977.
© Christian Schlötterer, Institut für Tierzucht und Genetik, Veterinärmedizinsche Universität Wien, 1999